All medicines are made of either small molecules or large molecules. Biologics are advanced therapeutic drugs made of living cells, sugars, proteins, tissues, or a mixture of these. Biosimilars as follow-on biologics are game-changers to reduce patient and payers’ health bills. Biosimilars are made of large molecules and living cells. They show similar pharmacokinetics (P.K.) and pharmacodynamics (P.D.) results identical to their reference listed biologics/drug (RLD). Biosimilars and sustainability go hand in hand.
The road to developing biosimilars is long and more complicated than developing generic drugs. However, all the efforts are worthwhile as biosimilars can increase the accessibility of advanced medications to more patients. This blog will discuss the exhaustive biosimilar approval pathways, growth drivers, and restraint factors that are fast-changing the healthcare scene worldwide.
Generic Drugs V/S Biosimilars
There are two widely available drugs, one either chemically synthesized Active Pharmaceutical Ingredients (APIs) or other biologically Active Pharmaceutical Ingredients (APIs). All chemically synthesized drugs have small molecules, which can be branded or generic. Conversely, biosimilars always use large molecules and have higher cynical similarity to their originator/innovator biologics.
A 5 year exclusivity period is granted to small molecule drugs that contain novel chemical entities that have never been used previously, either alone or in combination, by the FDA. In the E.U., an up to 10-year exclusivity period is provided to a generic medicinal Marketing Authorization Holder (MAH). Biologics, however, have a 12-year market exclusivity and eight-year data exclusivity period in the U.S. In the E.U., the biologics have 8+2+1 market exclusivity and eight years of data exclusivity.
Biosimilars reach the market after the exclusivity period in any market. Below, we share some core differences between small-molecule/single-molecule drugs and large-molecule biological/biosimilar drugs.
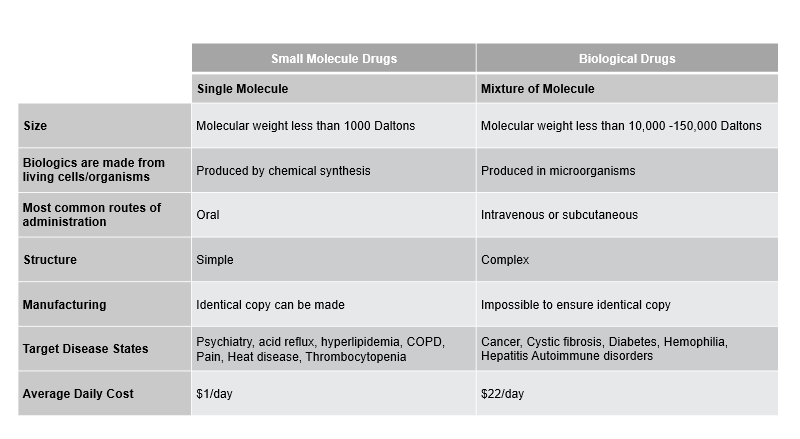
Figure 1: Key differences between Small Molecule Drugs and Biological Drugs.
Approval Pathways for Biosimilars in the U.S.
The United States introduced the first biosimilar in 2015, filgrastim-sndz, a granulocyte colony-stimulating factor injection that treats neutropenia.
The abbreviated 351 (a) or 351 (k) approval pathways for biosimilars in the U.S. are regulated by the FDA’s Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER). As biosimilars are not generics of the reference product, they often have some microheterogeneity due to the presence of large molecules and living cells. The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) simplified the road for developing generics and biosimilars.
With this act, undertakings in the biosimilar field don’t require one to include unnecessary clinical trials. Phase III clinical trials became unnecessary for biosimilar approvals post-keen PK/PD studies. However, it is imperative to demonstrate that the biosimilar must meet the quality/purity, efficacy/potency, and safety parameters.
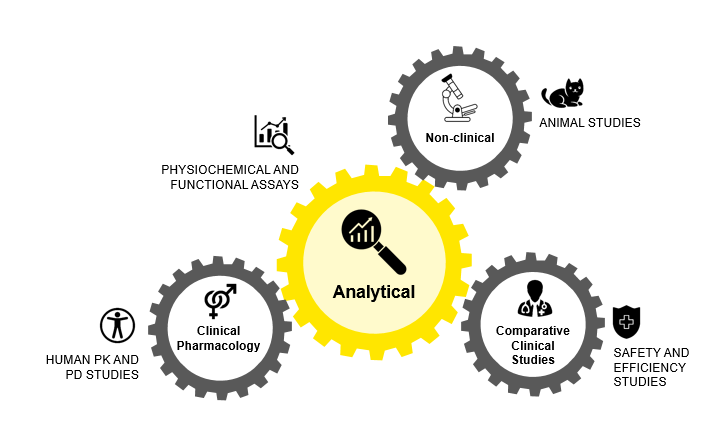
Figure 2: Approval Pathways for Biosimilars in U.S.
There are four interchangeable biosimilars approved by the FDA: Cyltezo (adalimumab-adbm), Cimerli (ranibizumab-eqrn), Rezvoglar (insulin glargine-aglr), and Semglee (insulin glargine-yfgn) which can be switched with other biosimilars in the same category without a prior prescription.
Biosimilars and Sustainability: Approval Pathways in E.U.
The first biosimilar Omnitrope (Somatropin), a human growth hormone, was approved by the E.U. in 2006. Likewise, the first biosimilar to receive approval in Japan was Somatropin in 2009. Aczicrit (epoetin lambda) was the first biosimilar to be approved in Australia in 2010. In contrast, in India, the first biosimilar (vaccine) was launched in 2000 for Hepatitis B.
In Europe, the European Medicines Agency (EMA) and the Committee for Medicinal Products for Human Use (CHMP) examine the process of marketing biosimilars. E.U. has four approval processes for biosimilars similar to generic drugs:
1. Centralised procedure
2. Decentralised procedure
3. Mutual recognition procedure
4. National procedure
The applicant has to submit the dossier according to Directive 2001/83/E.C.E.C. E.U., which also has scientific guidelines on biosimilars to develop these medicinal products. EMA and Heads of Medicines Agencies (HMA) state that biosimilars approved in the E.U. can be considered interchangeable, bringing more clarity to healthcare professionals, pharmacies, and patients to experience more ease from using biosimilars. E.U. has also made available key clinical data under the European Public Assessment Report (EPAR) program in reference to biosimilar trials so that such knowledge is freely available to biosimilar developers. This is done so that resources are not wasted in conducting previously approved tests.
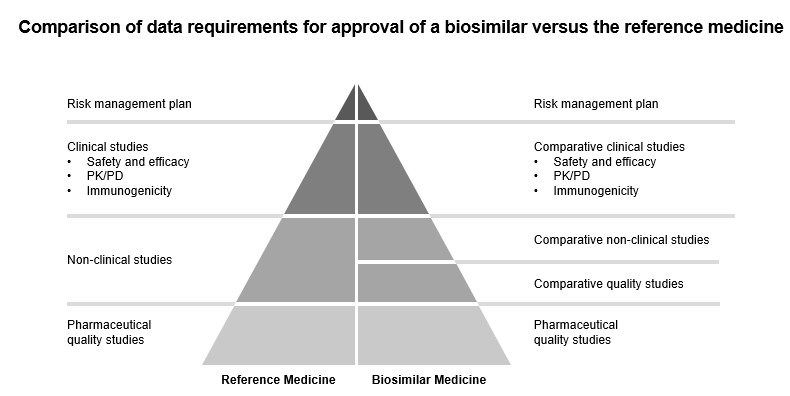
Figure 3: Comparison of data requirements for approval of a biosimilar versus the reference medicine
Immunogenicity of Biosimilars
All biologics are proteins (polypeptides), and they are segmented by types like microorganisms or mammalian cells, product types like monoclonal, recombinant proteins, antisense, and vaccines. In blood research and tissue advanced research, some biologics therapeutics can result in unwanted immune responses, such as the development of anti-drug antibodies against the fused FVIII, which is given to patients with hemophilia A.
As per the clinical observation, Black African folks were more likely to develop antibodies against factor VIII proteins than patients of European Caucasian descent. It has been suggested that a personalized approach to predict and avoid this kind of immune response to FVIII proteins can be done. This can be achieved by developing specific versions of genetically engineered therapeutic proteins. The researchers are using this knowledge and collaborating with the Center for Drug Evaluation and Research (CDER) to develop biosimilars that will be safe and effective.
Recombinant Proteins and Biosimilars
Human diseases are often caused by dysfunction of specific proteins. Medical biotechnology has used recombinant protein biotherapeutics to treat various illnesses and chronic conditions. The third generation of recombinant proteins is changing the biologics and biosimilar market with novel routes, new formulations, and increased safety and efficiency.
Major Pharma companies are developing their recombinant proteins as part of their larger portfolio, with more than 170 recombinant protein drugs available in the market. The recombinant non-glycosylated protein biosimilar market is expected to grow highly. Moreover, major companies like Sandoz International GmbH, Pflizer Inc., Celltrion Healthcare Co. Ltd., Samsung, Mylan N.V.N.V., Biocon, Amgen Inc., Dr. Reddy’s Laboratories Ltd., Stada Arzneimittel AG are working in Insulin, rHGH and Interferon biosimilars development.
Biosimilars and Sustainability: Exploring New Pharmacodynamics Research
The healthcare industry is looking to expedite research in biosimilars. One way to do this is by using pharmacodynamics (P.D.) biomarkers to demonstrate biosimilarity. Biosimilar sponsors’ ultimate goal is to establish the clinical similarity of their proposed medical product to its reference product.
It was found that P.K. evaluations are more common in most FDA-approved biosimilars than P.D. similarity data. According to this study, the P.D. biomarker that shows the biological product’s mechanism (s) of action (s) can potentially be more sensitive for detecting clinically meaningful differences between two products. Thus, biomarkers that were used as secondary and exploratory endpoints in new drug development programs play a key role in biosimilar programs.
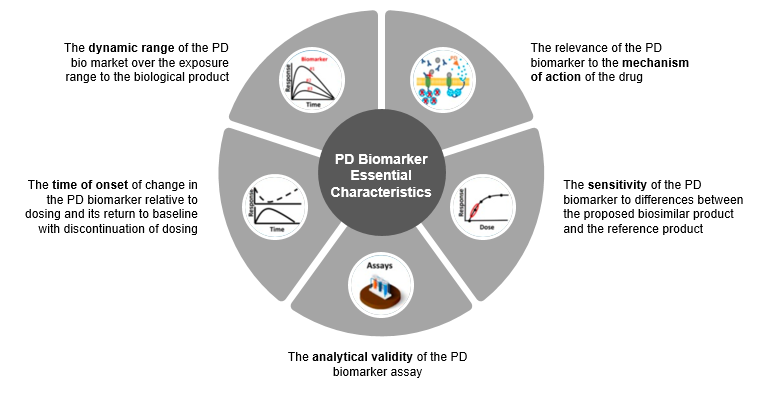
Figure 4: Five essential characteristics of P.D. (pharmacodynamic) biomarker for biosimilars as per FDA.
Role of Pharmacy Benefit Managers in Biosimilar Adoption
Global inflation has been high, close to 7.0% in 2023, and major economies are taking steps to reduce inflation. With the increase in chronic diseases and high health costs, many patients avoid taking their prescriptions, thus worsening their health conditions.
The three Pharmacy Benefit Managers (PBM), namely Express Scripts, CVS Caremark, and Optum Rx, own 89% of the market. PBM negotiates and contracts with all the various types of pharmacies on reimbursement, rebates, and prescription drug coverage terms in the U.S. Also, they are owned by insurance companies Cigna, Aetna, and UnitedHealth Group, respectively. Hence, the three largest PBMs are affiliated with health plans and pharmacies; therefore, the parent company controls up to three stages of the drug supply chain.
Pharmacy Benefit Managers must give biosimilars the right place to increase their adoption. Biosimilar market penetration depends on follow-up biologics market visibility and patient/member/provider preference for them. PBMs can better understand the cost-saving biosimilarity of these drugs with pharmacies, doctors, and hospitals. With increased support for biosimilars from PBMs, cost savings are likely to be realized. As a result, patients will benefit from these lower health costs.
Advanced Technologies in Biosimilar Research
Off-patent biologics development requires the latest and advanced technologies. Their development period must decrease by 30-40% compared to the current 7-10 years. Additionally, crystallography with synchrotron light, X-ray crystallography, mass spectrometry, spectroscopy, and other newer technologies like AI will more drastically change how biosimilars are built in the future.
Biosimilars and Sustainability: Impact
In the U.S., over nine blockbuster drugs are losing exclusivity in 2023, and more biosimilars will reach the market by 2026. The value of global biosimilars will triple by 2030 and reach 74 billion.
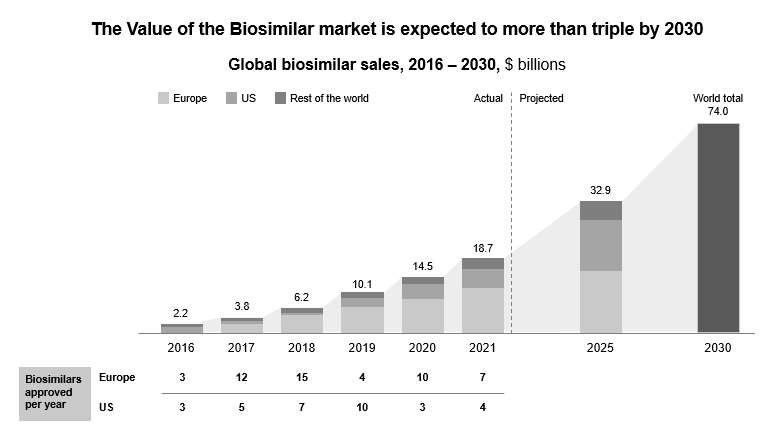
Figure 5: Biosimilar and sustainability market is expected to triple by 2030
Immunology, anti-diabetics, oncology, respiratory agents, antithrombotic, and multiple sclerosis form the leading biosimilar drug classes. Biosimilar sustainability improves the overall quality of healthcare delivery across the country. Globally, the biopharmaceutical industry, like all industries, needs more sustainable efforts. Companies developing Biologics Advanced Therapy Medicinal Products (ATMPs) and Biosimilar ATMPs need to focus on improving access to advanced therapies for one and all. There is a need to look at healthcare from a long-term perspective as today’s research becomes tomorrow’s medicines. As biosimilars have biological elements, these are less harmful to the water bodies and, hence, more sustainable.
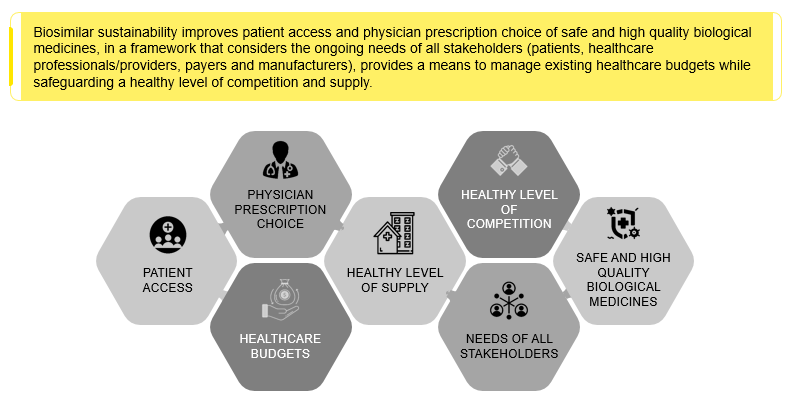
Figure 6: Biosimilars and Sustainability outlook involving all the stakeholders.
Further, the global savings from the biosimilar market will amount to $ 383 billion between 2023 and 2027. Moreover, the savings will be most visible by 2027 as key biologics like Ibrance lose market exclusivity.
Biosimilars and Sustainability: Conclusion
As we have seen, small-molecule generics of branded drugs have brought down the cost of medicines. With four and five follow-on biologics to originator drugs, we can see that Biosimilars will become more affordable. At the developer stage, the originator manufacturer, biosimilar sponsor, and respective authorization bodies should collaborate to address patent issues and strive to minimize unnecessary clinical trials. New technologies will continue apace the biosimilar market launches.